When uploading data to R, we sometimes worry about losing track of the data over time. This is because we save data in different folders according to various projects, and we might forget where we stored it. Additionally, if the file path changes, it can be difficult to upload the data directly and locate its current location.
Therefore, a better approach is to save the data as code, allowing us to access it directly when opening the R file where the code is stored. The most common method for converting data to code is by using dput(), but there are also other ways to achieve this in R. For more details, please check the post below.
■ How to convert an uploaded data to code in R?
Let’s talk in detail with an actual dataset.
if(!require(readr)) install.packages("readr")
library(readr)
github= "https://raw.githubusercontent.com/agronomy4future/raw_data_practice/main/fertilizer_treatment.csv"
dataA= data.frame(read_csv(url(github),show_col_types = FALSE))
print(head(dataA,5))
Genotype Block variable value
1 Genotype_A I Control 42.9
2 Genotype_A II Control 41.6
3 Genotype_A III Control 28.9
4 Genotype_A IV Control 30.8
5 Genotype_B I Control 53.3
.
.
.
Here is a dataset called dataA. If you upload this data from an Excel file on your PC, you need to save the file and specify the path every time you upload it to R. To avoid this, I want to save the dataset as code so that it can be directly stored in an R script and easily accessed without needing to reload the original file.
To achieve this, I used dput().
dput(dataA)
structure(list(Genotype = c("Genotype_A", "Genotype_A", "Genotype_A",
"Genotype_A", "Genotype_B", "Genotype_B", "Genotype_B", "Genotype_B",
"Genotype_C", "Genotype_C", "Genotype_C", "Genotype_C", "Genotype_D",
"Genotype_D", "Genotype_D", "Genotype_D", "Genotype_A", "Genotype_A",
"Genotype_A", "Genotype_A", "Genotype_B", "Genotype_B", "Genotype_B",
"Genotype_B", "Genotype_C", "Genotype_C", "Genotype_C", "Genotype_C",
"Genotype_D", "Genotype_D", "Genotype_D", "Genotype_D", "Genotype_A",
"Genotype_A", "Genotype_A", "Genotype_A", "Genotype_B", "Genotype_B",
"Genotype_B", "Genotype_B", "Genotype_C", "Genotype_C", "Genotype_C",
"Genotype_C", "Genotype_D", "Genotype_D", "Genotype_D", "Genotype_D",
"Genotype_A", "Genotype_A", "Genotype_A", "Genotype_A", "Genotype_B",
"Genotype_B", "Genotype_B", "Genotype_B", "Genotype_C", "Genotype_C",
"Genotype_C", "Genotype_C", "Genotype_D", "Genotype_D", "Genotype_D",
"Genotype_D"), Block = c("I", "II", "III", "IV", "I", "II", "III",
"IV", "I", "II", "III", "IV", "I", "II", "III", "IV", "I", "II",
"III", "IV", "I", "II", "III", "IV", "I", "II", "III", "IV",
"I", "II", "III", "IV", "I", "II", "III", "IV", "I", "II", "III",
"IV", "I", "II", "III", "IV", "I", "II", "III", "IV", "I", "II",
"III", "IV", "I", "II", "III", "IV", "I", "II", "III", "IV",
"I", "II", "III", "IV"), variable = c("Control", "Control", "Control",
"Control", "Control", "Control", "Control", "Control", "Control",
"Control", "Control", "Control", "Control", "Control", "Control",
"Control", "Fertilizer1", "Fertilizer1", "Fertilizer1", "Fertilizer1",
"Fertilizer1", "Fertilizer1", "Fertilizer1", "Fertilizer1", "Fertilizer1",
"Fertilizer1", "Fertilizer1", "Fertilizer1", "Fertilizer1", "Fertilizer1",
"Fertilizer1", "Fertilizer1", "Fertilizer2", "Fertilizer2", "Fertilizer2",
"Fertilizer2", "Fertilizer2", "Fertilizer2", "Fertilizer2", "Fertilizer2",
"Fertilizer2", "Fertilizer2", "Fertilizer2", "Fertilizer2", "Fertilizer2",
"Fertilizer2", "Fertilizer2", "Fertilizer2", "Fertilizer3", "Fertilizer3",
"Fertilizer3", "Fertilizer3", "Fertilizer3", "Fertilizer3", "Fertilizer3",
"Fertilizer3", "Fertilizer3", "Fertilizer3", "Fertilizer3", "Fertilizer3",
"Fertilizer3", "Fertilizer3", "Fertilizer3", "Fertilizer3"),
value = c(42.9, 41.6, 28.9, 30.8, 53.3, 69.6, 45.4, 35.1,
62.3, 58.5, 44.6, 50.3, 75.4, 65.6, 54, 52.7, 53.8, 58.5,
43.9, 46.3, 57.6, 69.6, 42.4, 51.9, 63.4, 50.4, 45, 46.7,
70.3, 67.3, 57.6, 58.5, 49.5, 53.8, 40.7, 39.4, 59.8, 65.8,
41.4, 45.4, 64.5, 46.1, 62.6, 50.3, 68.8, 65.3, 45.6, 51,
44.4, 41.8, 28.3, 34.7, 64.1, 57.4, 44.1, 51.6, 63.6, 56.1,
52.7, 51.8, 71.6, 69.4, 56.6, 47.4)), class = "data.frame", row.names = c(NA,
-64L))
However, the outputted code is spread across multiple lines, taking up a lot of space. When I copy and paste it into an R script or Google Colab, it looks messy and unorganized.

■ datazip() package
Therefore, I want a simple one-line code format. The datazip() package I developed is a useful tool for converting data into a single-line code format.
First, let’s import the package.
if(!require(remotes)) install.packages("remotes")
if (!requireNamespace("datazip", quietly = TRUE)) {
remotes::install_github("agronomy4future/datazip", force= TRUE)
}
library(remotes)
library(datazip)
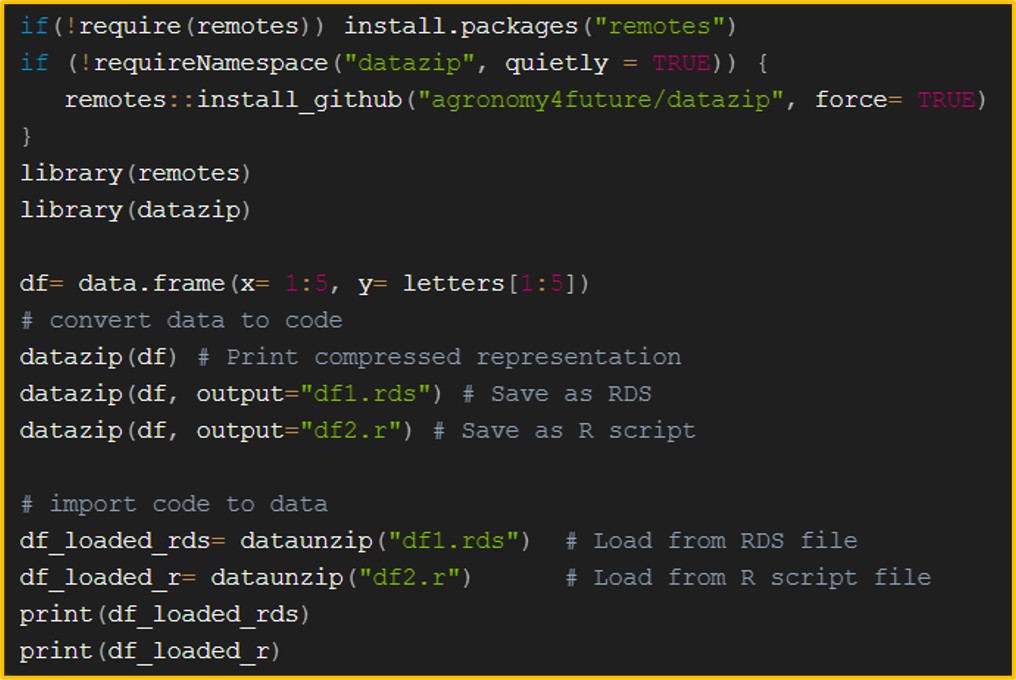
If you run ?datazip, you can view its description.
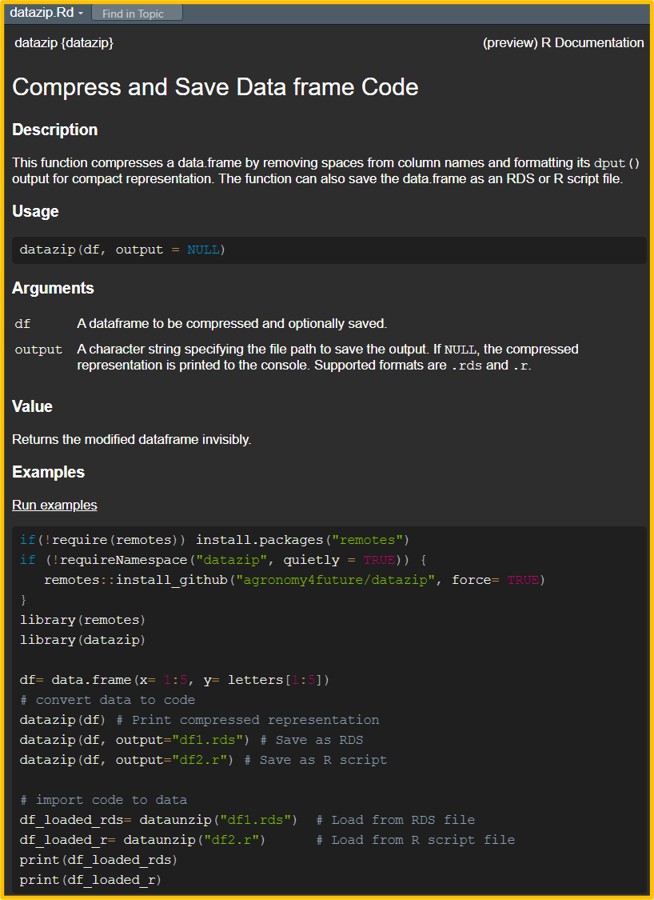
First, I’ll convert the data into a single-line code.
datazip(dataA)
structure(list(Genotype=c("Genotype_A","Genotype_A","Genotype_A","Genotype_A","Genotype_B","Genotype_B","Genotype_B","Genotype_B","Genotype_C","Genotype_C","Genotype_C","Genotype_C","Genotype_D","Genotype_D","Genotype_D","Genotype_D","Genotype_A","Genotype_A","Genotype_A","Genotype_A","Genotype_B","Genotype_B","Genotype_B","Genotype_B","Genotype_C","Genotype_C","Genotype_C","Genotype_C","Genotype_D","Genotype_D","Genotype_D","Genotype_D","Genotype_A","Genotype_A","Genotype_A","Genotype_A","Genotype_B","Genotype_B","Genotype_B","Genotype_B","Genotype_C","Genotype_C","Genotype_C","Genotype_C","Genotype_D","Genotype_D","Genotype_D","Genotype_D","Genotype_A","Genotype_A","Genotype_A","Genotype_A","Genotype_B","Genotype_B","Genotype_B","Genotype_B","Genotype_C","Genotype_C","Genotype_C","Genotype_C","Genotype_D","Genotype_D","Genotype_D","Genotype_D"),Block=c("I","II","III","IV","I","II","III","IV","I","II","III","IV","I","II","III","IV","I","II","III","IV","I","II","III","IV","I","II","III","IV","I","II","III","IV","I","II","III","IV","I","II","III","IV","I","II","III","IV","I","II","III","IV","I","II","III","IV","I","II","III","IV","I","II","III","IV","I","II","III","IV"),variable=c("Control","Control","Control","Control","Control","Control","Control","Control","Control","Control","Control","Control","Control","Control","Control","Control","Fertilizer1","Fertilizer1","Fertilizer1","Fertilizer1","Fertilizer1","Fertilizer1","Fertilizer1","Fertilizer1","Fertilizer1","Fertilizer1","Fertilizer1","Fertilizer1","Fertilizer1","Fertilizer1","Fertilizer1","Fertilizer1","Fertilizer2","Fertilizer2","Fertilizer2","Fertilizer2","Fertilizer2","Fertilizer2","Fertilizer2","Fertilizer2","Fertilizer2","Fertilizer2","Fertilizer2","Fertilizer2","Fertilizer2","Fertilizer2","Fertilizer2","Fertilizer2","Fertilizer3","Fertilizer3","Fertilizer3","Fertilizer3","Fertilizer3","Fertilizer3","Fertilizer3","Fertilizer3","Fertilizer3","Fertilizer3","Fertilizer3","Fertilizer3","Fertilizer3","Fertilizer3","Fertilizer3","Fertilizer3"),value=c(42.9,41.6,28.9,30.8,53.3,69.6,45.4,35.1,62.3,58.5,44.6,50.3,75.4,65.6,54,52.7,53.8,58.5,43.9,46.3,57.6,69.6,42.4,51.9,63.4,50.4,45,46.7,70.3,67.3,57.6,58.5,49.5,53.8,40.7,39.4,59.8,65.8,41.4,45.4,64.5,46.1,62.6,50.3,68.8,65.3,45.6,51,44.4,41.8,28.3,34.7,64.1,57.4,44.1,51.6,63.6,56.1,52.7,51.8,71.6,69.4,56.6,47.4)),class="data.frame",row.names=c(NA,-64L))
The code is generated as a single line. When copying and pasting it into an R script or Google Colab, it takes up much less space.
Eventually, this code enables the simple conversion of data into a single-line code, making it easy to save as a script.
However, there is one limitation—saving large datasets as code can be challenging. Let’s upload a large dataset to explore this issue.
if(!require(readr)) install.packages("readr")
library(readr)
github="https://raw.githubusercontent.com/agronomy4future/raw_data_practice/main/wheat_grains_data_training.csv"
dataB=data.frame(read_csv(url(github),show_col_types= FALSE))
print(tail(dataB,5))
Field Genotype Block fungicide planting_date fertilizer Shoot Length.mm. Width.mm.
96315 South Peele III No early N/A Tillers 5.951 2.987
96316 South Peele III No early N/A Tillers 5.614 2.687
96317 South Peele III No early N/A Tillers 5.674 2.210
96318 South Peele II No late N/A Tillers 6.041 2.138
96319 South Peele II No late N/A Tillers 6.041 2.138
Area.mm2.
96315 13.687
96316 11.058
96317 9.154
96318 18.092
96319 18.092
This dataset contains 96,319 rows, and when converted into code, it becomes too large to display in an R script.
Let’s try it!
datazip(dataB)
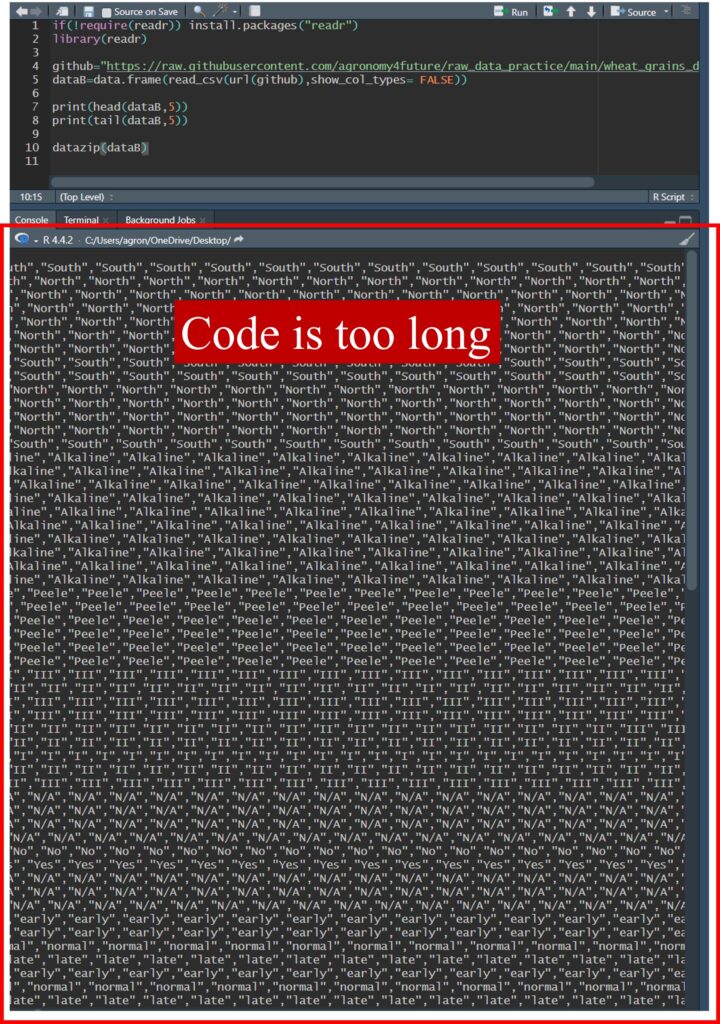
The generated code is too long to copy and paste, making it difficult to scroll through in the R console. There is no practical benefit to saving such a large code block in your R script. In this case, a better approach is to export the code to R file instead.
So, I’ll use the code below to designate that the code will be saved as an .r file, with "output="data_name.r". I exported this code to my PC ("C:/Users/Desktop") as an R file named dataB_output.
setwd("C:/Users/Desktop") # set up the pathway to save the file
datazip(dataB, <mark style="background-color:rgba(0, 0, 0, 0)" class="has-inline-color has-vivid-red-color">output="dataB_output.r"</mark>)
After the code is downloaded to my PC, when I open the file, you might see the following message:
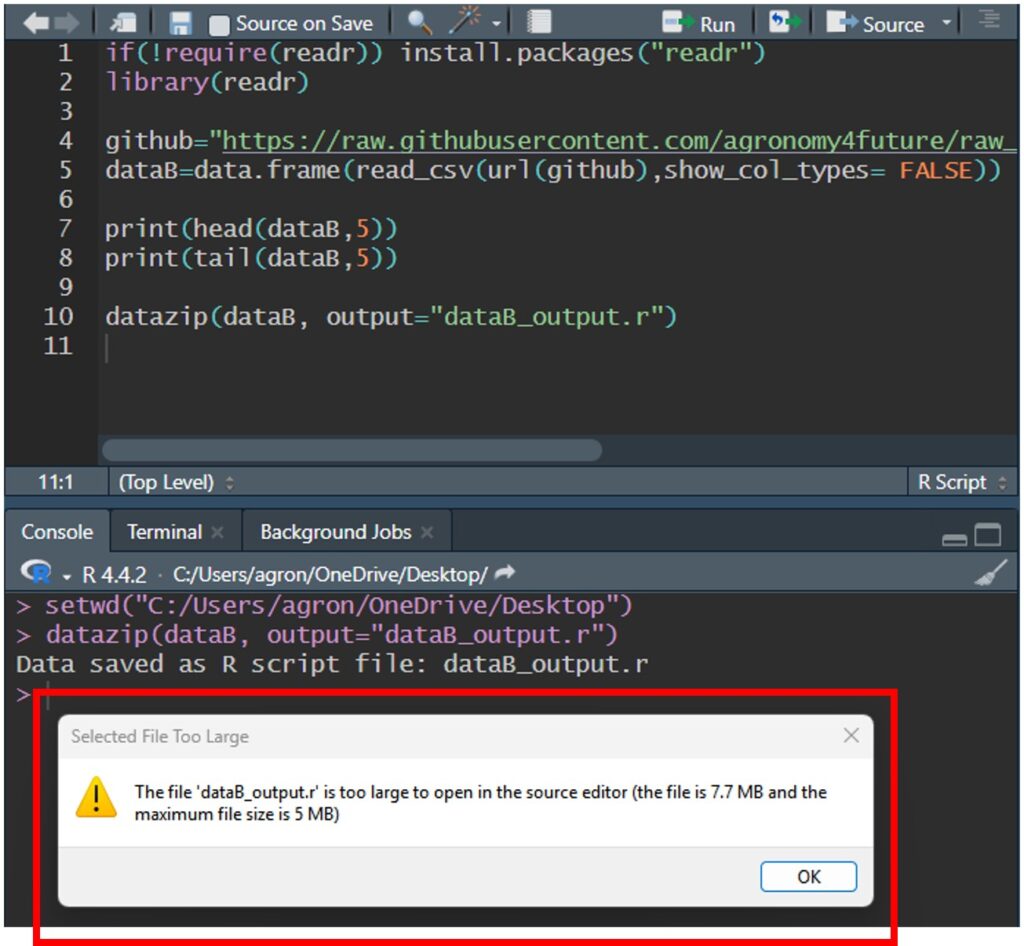
This code is too large to open in the source editor. However, dataB_output.r is still saved as code.
Later, if I want to open this code as a data table, we can use the R package dataunzip(), which I developed as a counterpart to datazip().
setwd("C:/Users/Desktop") # set up the pathway to import the file
dataB_recovered= dataunzip("dataB_output.r")
print(tail(dataB_recovered, 5))
Field Genotype Block fungicide planting_date fertilizer
96315 South Peele III No early N/A
96316 South Peele III No early N/A
96317 South Peele III No early N/A
96318 South Peele II No late N/A
96319 South Peele II No late N/A
Shoot Length.mm. Width.mm. Area.mm2.
96315 Tillers 5.951 2.987 13.687
96316 Tillers 5.614 2.687 11.058
96317 Tillers 5.674 2.210 9.154
96318 Tillers 6.041 2.138 18.092
96319 Tillers 6.041 2.138 18.092
.
.
.
Now the code is recovered as a data table again! From now on, you can save your data as code instead of as an Excel file, which might be modified when opened or moved.
If you want to save it in .rds format (not as code, but as a single R object), you can use the code below.
# to save data as .rds
datazip(df, output="dataB_output.rds")
# to import it to R as data frame
dataB_recovered= dataunzip("dataB_output.rds")
print(tail(dataB_recovered, 5))
This datazip() and dataunzip() package allows for the simple conversion of data into code with a single line, making it easy to save as a script.
Github: https://github.com/agronomy4future/datazip
We aim to develop open-source code for agronomy ([email protected])
© 2022 – 2025 https://agronomy4future.com – All Rights Reserved.
Last Updated: 06/03/2025